
BioML Challenge 2024: Bits to Binders
Published:
CAR-T cell therapy is a cancer treatment where a patient’s T cells are genetically modified to fight cancer. The process involves extracting T cells from the patient, engineering them to express a Chimeric Antigen Receptor (CAR) specifically targeting cancer cells, and reintroducing these modified cells into the patient’s body. Once inside the patient’s body, the CAR-T cells recognize and attack the cancer cells, helping to eliminate the tumor. An open question in the field of CAR-T cell therapy is how to design effective antigen-binding domains that can specifically target cancer antigens and trigger a robust immune response.
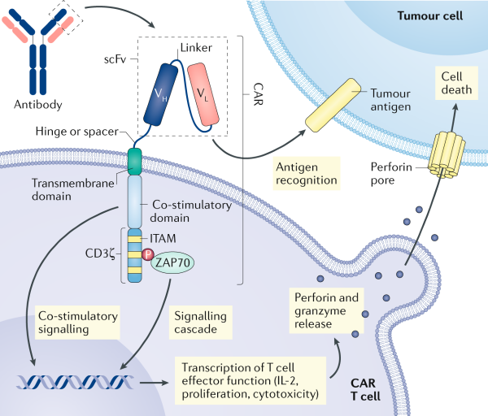
Larson et al., 2021, Nature Reviews Cancer
This project aims to design binding domains for a Chimeric Antigen Receptor (CAR) that will engage the extracellular region of cancer antigen CD20. The goal of this project is to gain practical experience in the binder design workflow, implement a standardized pipeline, and explore how different, SOTA generative models play a critical role in the design process. Furthermore, the project will apply advanced AI tools to the design challenge, observing both their capabilities and limitations in generating functional CAR antigen binders.
I want to thank BioML for hosting this awesome challenge. I also appreciate Lance Liu, Zhaoqian Su, Prof. Rocco Morreti from Meiler Lab and Prof. Tyler Derr from NDS Lab for their valuable guidance and help on this project. The code and technical report will be available soon! Here are some examples of the top binder designs from our submission.
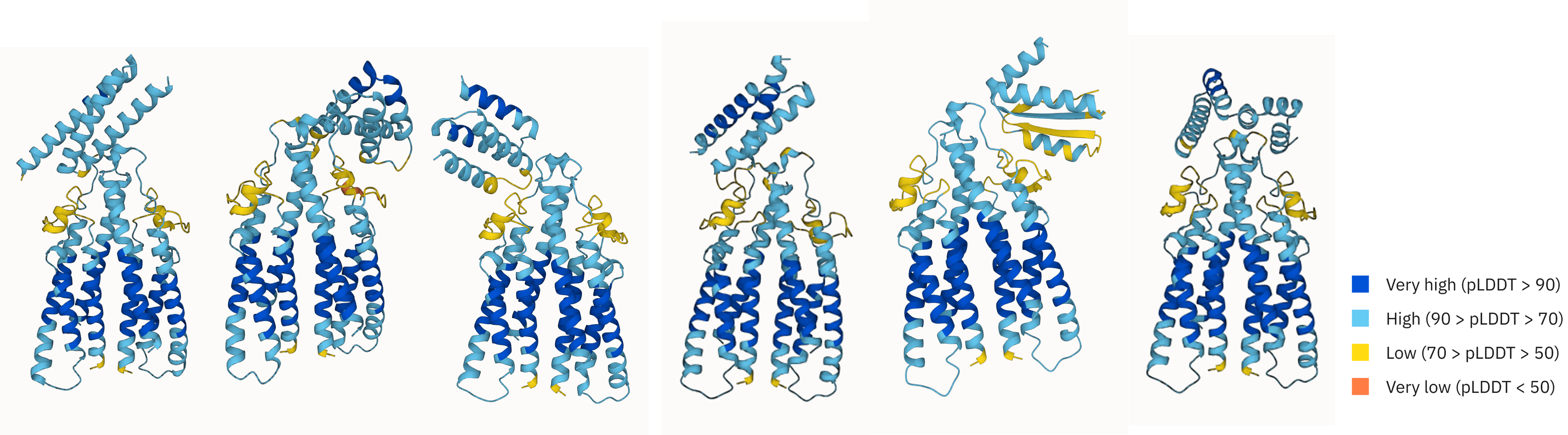
Top binder designs with diverse structures
Method Overview
I employed a standard pipeline encompassing backbone generation, inverse folding, and filtering to tackle the challenge. For binder design with the RFdiffusion model, our focus was on generating binder backbones that specifically target epitope 3 of CD20, located in the extracellular region. A diverse subset of these backbones was selected for inverse folding using ProteinMPNN, producing multiple sequence designs for each backbone. AlphaFold2 was then used to validate the 3D structures of the sequence designs by comparing the RMSD score to the original backbone. Finally, the Chai-1 model predicted the complex formed between CD20 and the filtered binder sequences. After excluding complex predictions that failed to meet the correct binding site and extracellular constraints, I ranked the binder sequences based on their ipTM scores output by Chai-1.
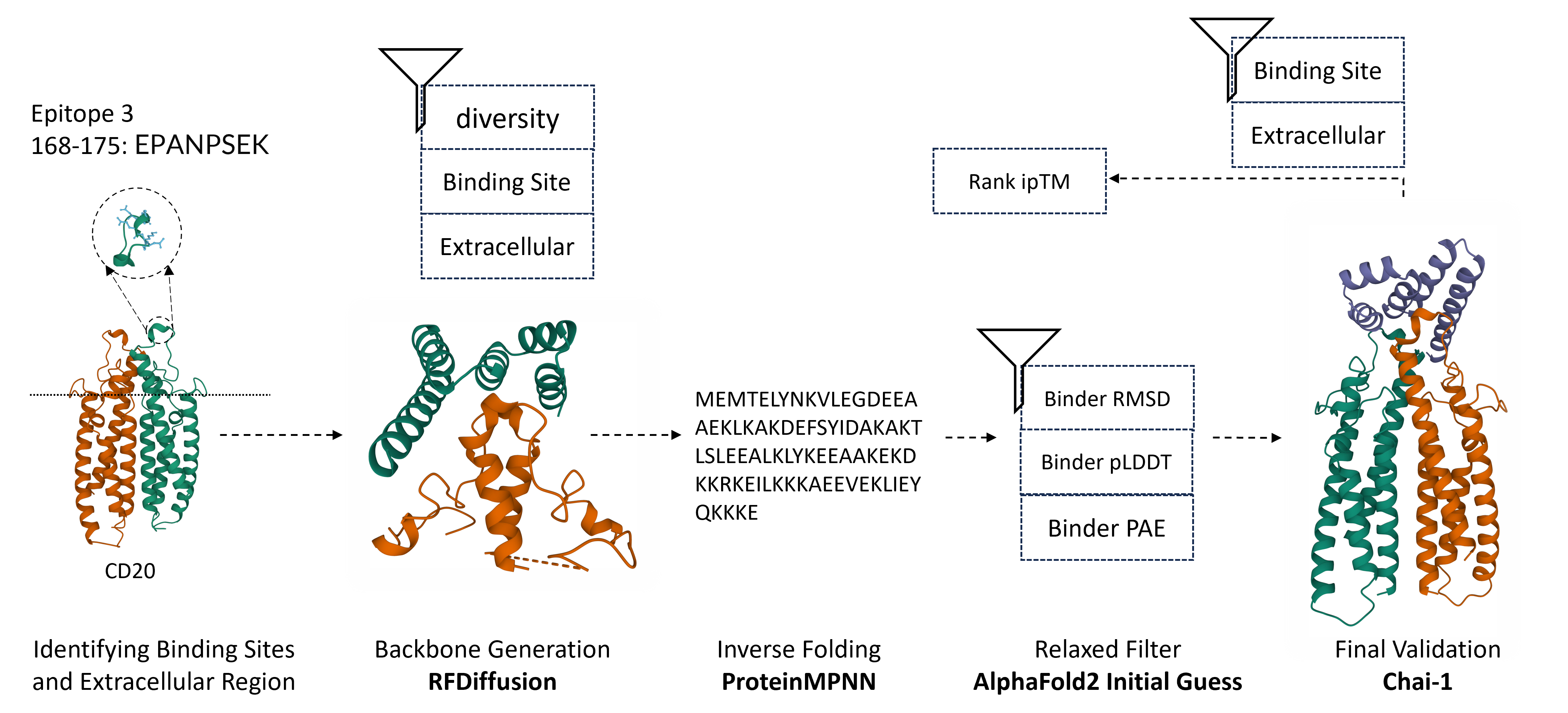
The pipeline of the binder design
Understanding the Antigen CD20
Before starting the binder design, I need to fully understand our antigen target, CD20. The best approach is to visualize its 3D structure and identify the extracellular region as Ill as the potential binding site within it. I can download CD20’s PDB file from uniprot and use PyMOL to visualize the structure after removing unwanted ligands and proteins. From the uniprot, I learned that CD20’s Epitope 3: 168-175 (Sequence: EPANPSEK) was recognized by antibodies, including Rituximab, through experiments. Then, I should keep in mind to design binders that interact with these amino acids and lie in the extracellular region. Here is a image that roughly circles the potential binding site of CD20 and segments the extracellular and the remaining regions.
Backbone Generation via RFDiffusion
RFDiffusion is a computational tool used to design protein binders by leveraging diffusion models to generate novel protein structures. It plays a crucial role in binder design by sampling potential binder conformations that can interact specifically with a target protein. However, RFDiffusion can only generate the backbone of the binder without sequence information, which means further steps are needed to determine the amino acid sequence of the binder. For this challenge, I configured RFDiffusion with the following considerations: (1) extracellular region, (2) binding site, and (3) diversity of the designed binder.
By experimenting with RFDiffusion, I found truncating the target input and only keeping the extracellular part could prompt the model to generate binders in the extracellular region. Besides, since RFDiffusion scales in runtime as \(O(N^2)\) where \(N\) is the number of amino acids in the system, truncating the protein target significantly reduced the inference run time for the large-scale design.
RFDiffusion allows the user to define hotspots, which control the binder to interact with a specific site on the protein target. After carefully examining Epitope 3 mentioned earlier, and the complex including CD20 and existing antibodies, I defined a few hotspots that allow diverse binding scenarios.
An example of Binder Backbone (green) with truncated CD20 (blue) output by RFDiffusion
To encourage the diversity of our designed binders, I adopted the complex_beta checkpoint of RFDiffusion, which can generate a greater diversity of topologies compared to the base model. Empirically, the base model could only generate simple helical binders that share high structural similarity. Additionally, I tuned noise_scale_ca and noise_scale_frame with values of 0, 0.5, and 1. These two hyperparameters indicate the noise added to the position of \(C_{\alpha}\) and the frame (translation and rotation) during inference. Reducing the noise can improve the quality of binder designs, but at the expense of diversity. I generated approximately 10,000 binder backbones across three different noise levels and only kept ones that interacting around the hotspots. Then, I refined the selection by clustering to retain only a subset of diverse topologies. Specifically, I aligned all binder backbones, calculated an RMSD matrix, and performed hierarchical clustering with a cutoff of \(\pu{5 Å}\). From each resulting cluster, a representative binder was selected. This process yielded 1,500 diverse binder backbones for the inverse folding step.
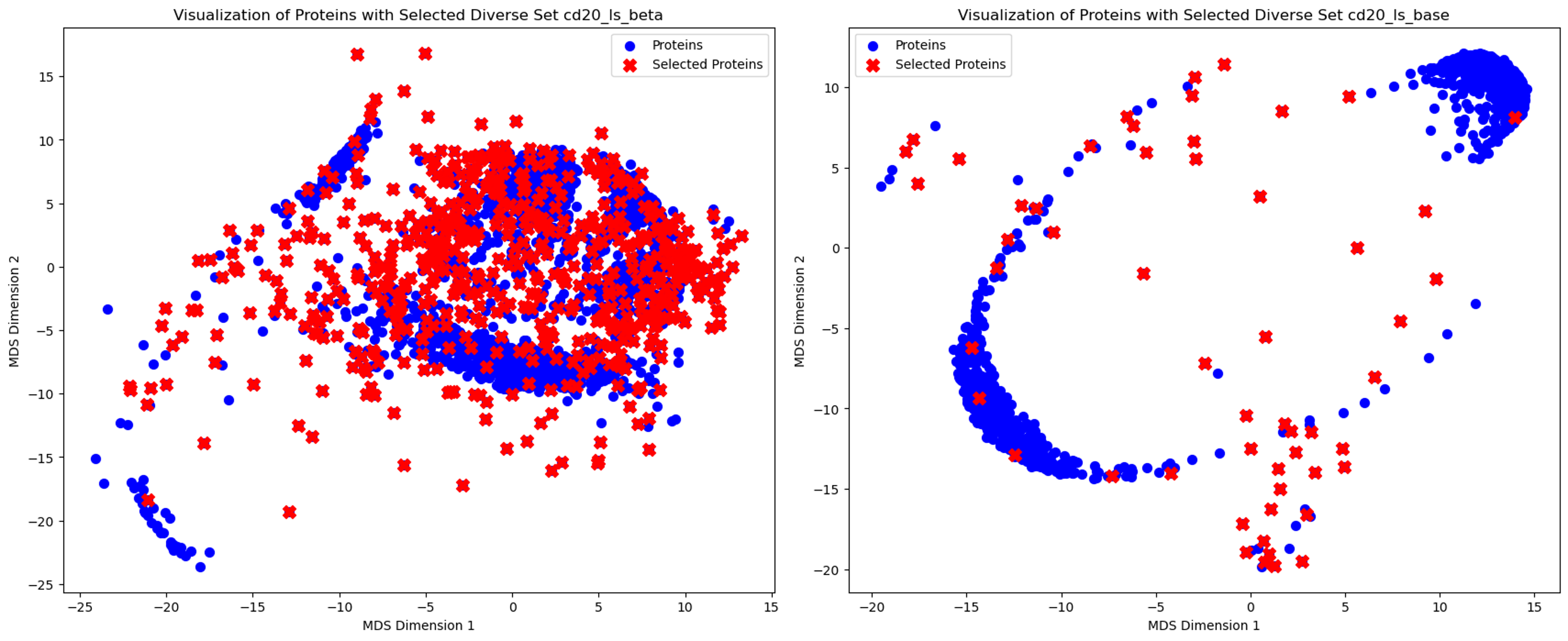
Comparision betIen beta and base model checkpoints: by projecting the RMSD in 2D space, I can see the beta model on the left can generate more structural diverse binders, compared to the base model on the right. With the same cutoff value, more clusters (red cross) are derived from binders designed by the beta model.
Inverse Folding via ProteinMPNN
Inverse folding is a process in which a protein backbone structure is used to predict a corresponding amino acid sequence that would fold into that specific structure. ProteinMPNN is a deep learning-based model specifically developed for the task of inverse protein folding, where the goal is to predict an amino acid sequence that will fold into a given 3D protein structure. It uses a message-passing neural network to model the relationships and interactions between residues in a protein, allowing it to efficiently design sequences that are highly compatible with the input backbone. ProteinMPNN works by optimizing the sequence to achieve favorable intramolecular interactions, ensuring stability with the provided structure. Following the pipeline described in the RFDiffusion paper, I employed ProteinMPNN-FastRelax Protocol to design our 5 sequences for each filtered backbone.
An example of predicting the sequence from the binder backbone
In addition to ProteinMPNN, there are other methods for inverse folding, such as LigandMPNN and ESM-IF1. These models can also be used for this challenge, but I sticked with ProteinMPNN since its output is reasonable .
Relaxed Filtering via AlphaFold2 Initial Guess
I initially attempted to filter binder designs using AlphaFold2 Initial Guess, retaining only those with pae_interaction < 10, as described in the RFDiffusion paper. However, I found that AlphaFold2 Initial Guess was unable to predict successful binding between all sequences and CD20, with pae_interaction values often around 25. Therefore, we decided to rely solely on the metrics related to the binder output by AlphaFold2 (Binder RMSD, Binder PAE, and Binder pLDDT) to apply a more relaxed filtering for the sequences.
Binder RMSD: Measures how much the predicted structure deviates from the original backbone, indicating the level of structural similarity. I set a threshold of 2 Å to filter out designs with significant deviations.
Binder PAE: Represents the global confidence of AlphaFold2’s prediction by evaluating the accuracy of distance predictions between residues. A threshold of 7.5 Å was used to ensure sufficient global reliability in the predictions.
Binder pLDDT: Provides a local confidence score for each residue in the predicted structure, indicating how well the model thinks that specific region is predicted. I filtered designs to only keep those with a pLDDT score of at least 85, suggesting ok reliability in local structure prediction.
Eventually, I got 3,097 sequences associated with 1,007 unique backbones ready for the final Validation.
Final Complex Validation via Chai-1
To bypass the failure of AlphaFold2 Initial Guess, I tried AlphaFold-multimer and Chai-1 to predict the protein-binder complex.
AlphaFold-Multimer is an extension of AlphaFold2 designed to predict the structures of protein complexes involving multiple interacting chains, such as protein-protein interactions, antibody-antigen complexes, or homomeric and heteromeric assemblies. It builds upon the core architecture of AlphaFold2 but adds specific features to better handle the challenges of modeling multiple sequences interacting within a complex. One key aspect of AlphaFold-Multimer is the use of multiple sequence alignments (MSA) to gather evolutionary information for each chain in the complex. MSAs are used to identify conserved sequences and co-evolutionary patterns, which provide crucial insight into potential interactions between different proteins. By combining the MSAs of each chain, AlphaFold-Multimer can better understand and predict the inter-chain contacts. However, to achieve large-scale inference with AlphaFold-Multimer, I had to download the entire MSA dataset (~ 3GB), but I encountered difficulties doing this on my lab’s cluster.
Finally, I decided to use Chai-1, “a multi-modal foundation model for molecular structure prediction that performs at the state-of-the-art across a variety of tasks relevant to drug discovery,” which was released in September, 2024. The Chai Discovery team claims that Chai-1 excels in tasks such as protein-ligand and multimer prediction. Chai-1 can predict biopolymer structures from raw sequences and chemical inputs, and can also incorporate experimental constraints (e.g., epitope mapping or cross-linking data) for more accurate complex predictions. While Chai-1 performs best with MSAs, it also provides strong predictions in single-sequence mode, outperforming ESMFold and even AF-Multimer 2.3 in certain cases. However, the model only supports protein sequences in FASTA format as input now.

An example of Chai-1 prediction
I evaluated all the filtered sequences using Chai-1, focusing primarily on the PTM and IPTM scores for the predicted protein-binder complexes.
The PTM score reflects how well Chai-1 predicted the overall structure of the complex, with most values above 0.5.
The IPTM score indicates the accuracy of the predicted relative positions of the subunits forming the protein-protein complex, where the best results are above 0.7, although most top predictions fell between 0.55 and 0.6.
Then, I filtered the predicted complexes to ensure interaction at the correct binding site and ensured each binder sequence has a unique backbone. At last, I visually examined some sampled designs and submitted the top 500 designs based on their IPTM scores.
Reference
[1] Watson, J.L., Juergens, D., Bennett, N.R. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023). https://doi.org/10.1038/s41586-023-06415-8
[2] Bennett, N.R., Coventry, B., Goreshnik, I. et al. Improving de novo protein binder design with deep learning. Nat Commun 14, 2625 (2023). https://doi.org/10.1038/s41467-023-38328-5
[3] Chai Discovery team. (n.d.). Chai-1 technical report. https://chaiassets.com/chai-1/paper/technical_report_v1.pdf
[4] Jumper, John, et al. “Highly accurate protein structure prediction with AlphaFold.” nature 596.7873 (2021): 583-589.